

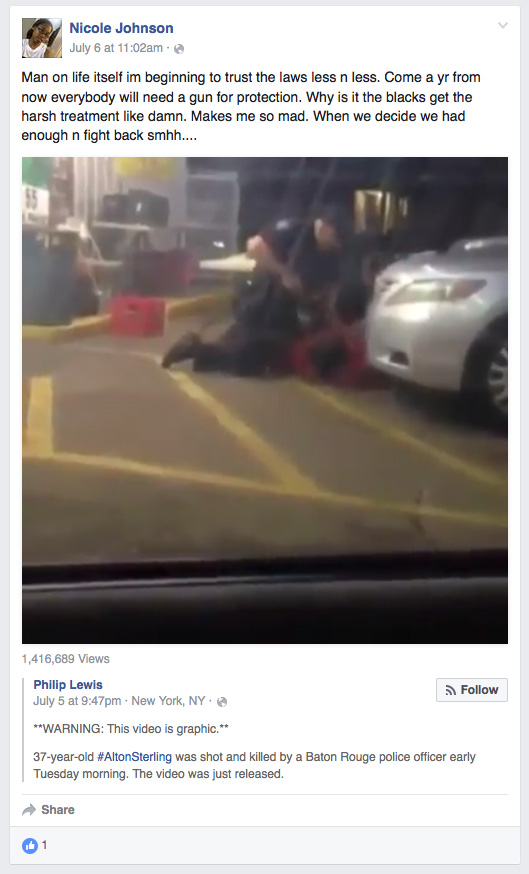
But let's get to the whiteness of whiteness! What do I mean? I mean these people are so absorbed in their mythology of whiteness they don't have a clue how damaging their whiteness is to themselves and others, therefore they are qualified for admission to the mental institution. They condemn any and everything Black people express in our attempt to communicate with them our feelings of humanity. We say Black Lives Matter and they know Black lives don't matter to them for they treat their dogs better than they treat us, so BLM means nothing to them thus they ridicule it and wish we would dispense the idea that our lives matter, for all lives matter in their sick minds while the news shows them nightly how little Black lives matter when pigs kill our men in front of our women and children, and often kill our children and women. And then they are bewildered why we cry Black Lives Matter. Of course, Baldwin said they live in an airless room and furthermore the idea of white supremacy has led them to rationalizations so fantastic it approaches the pathological!
They kill us at the drop of a hat, and then when we kill them they are utterly astounded that we would have the nerve to harm precious white flesh, and yet they will flip the subject to why we kill each other as if they haven't taught us to hate each other in the same manner they hate us. Every institution in American society suggests we must hate ourselves and love them. Every image of a woman is that of a white woman or a almost white Black woman. One need only look at the Rap videos to see how often a Black skinned woman is projected as the object of beauty, even though they know and we know the Black woman's body is the true standard of beauty since the beginning of time.
So our women are brainwashed to hate their black skin from Africa to Jamaica--bleaching cream is imported into Africa by the tons so women can emulate the European standard of beauty, and the same is true in India and China. Yes, White lunacy is global and the addiction to white lunacy is global. As is taught in drug and alcohol recovery, addiction is cunning and vile. One can have a relapse at the drop of a hat, the slip of a tongue can reveal the residue of white lunacy even while the addict claims recovery.
The solution to recovery from the addiction to the whiteness of whiteness or white lunacy is long term recovery to engender neuroplasticity that will allow the brain cells to change due to a new environment.
The more we try to make them part of the human family, the more they reveal themselves as part of the animal family of beasts and predators of the worse jungle variety. They want us to stop killing each other but who taught us to kill? They did and as we speak they are killing around the world for nothing. They cannot tell us why they are still killing in Iraq, Afghanistan, Syria, Yemen, Somalia, etc.
For that matter, can they tell us why they kill a human being for selling single cigarettes, CDs, DVDs,
a defective tail light, signal light, for going to the store for a soda, for being mentally ill? Why would you kill the mentally ill? It is because you are addicted to murder under the color of law. We cannot have a trillion dollar military budget to kill around the world, including a president who checks off a murder list weekly, then expect no blow back? What did James Baldwin tell you, "The murder of my child will not make your child safe!"
--Marvin X, How to Recover from the Addiction to White Supremacy, Black Bird Press, Berkeley CA.

Neuroplasticity
NEUROPLASTICITY
Information in the brain is transmitted from neuron to neuron through specialized connections called synapses. A synapse between two neurons is made up of presynaptic and postsynaptic terminals, which are separated by a synaptic cleft. The presynaptic terminal is filled with small vesicles containing chemical neurotransmitters, and the postsynaptic terminal consists of receptors specific for these neurochemicals. Neurons carry information in the form of an electrical impulse called an action potential that is initiated at the cell body and travels down the axon. At the synapse, an action potential causes the voltage-dependent release of neurotransmitter-filled vesicles, thereby converting an electrical impulse into a chemical signal. Neurotransmitters diffuse across the synaptic cleft, where they bind to receptors and generate an electrical signal in the postsynaptic neuron. The postsynaptic cell will then, in turn, fire an action potential if the sum of all its synapses reaches an electrical threshold for firing. Since a neuron can receive synapses from many different presynaptic cells, each cell is able to integrate information from varied sources before passing along the information in the form of an electrical code. The ability of neurons to modify the strength of existing synapses, as well as form new synaptic connections, is called neuroplasticity. It is believed that neuroplasticity may be the underlying cellular mechanism for the brain's ability to encode information during learning. In theory, this is how information is stored as memory.Defined in this way, neuroplasticity includes changes in strength of mature synaptic connections, as well as the formation and elimination of synapses in adult and developing brains. This encompasses a vast field of research, and similar processes may also occur at peripheral synapses, where much of the pioneering studies on synaptic transmission first took place. In addition, neuroplasticity includes the regrowth (or sprouting) of new synaptic connections following central nervous system injury; following stroke, for example.
The notion that the brain can store information by modifying synaptic connections is not a new one. In fact, Santiago Ramon y Cajal (a founder of modern neuroscience) expressed this theory in 1894, three years before Charles Sherrington coined the term synapse to describe the connections made between neurons. In the late 1940s the neuroplasticity model was advanced by Jerzy Konorski, who used the word plasticity to describe "permanent functional transformations," and Donald Hebb, who ascribed testable physiologic characteristics to synaptic plasticity. However, experimental evidence that synapses are capable of long-lasting changes in synaptic strength did not come until the early 1970s, when Timothy Bliss and Terry Lomo described an increase in the synaptic strength of neurons in the mammalian hippocampus (a region of the brain critical for some forms of memory) following electrical stimulation. They termed this increase long-lasting potentiation, now referred to as long-term potentiation (LTP).
Changes in synaptic strength proved to be bidirectionally modifiable (they increase and decrease in strength) as Serena Dudek and Mark Bear first demonstrated in 1992 by recording activity-driven, long-term depression (LTD) in the hippocampus. The evidence that learning and memory are based on these long-lasting changes in synaptic strength is substantial, but still incomplete. However, defining the molecular constituents in the mechanistic pathway leading from synaptic activity to plasticity continues to strengthen the evidence linking neuroplasticity with learning and memory. In addition, resolving the molecular mechanisms underlying synaptic modification should lead to targets for clinical intervention in eliminating age-related memory loss or synaptic loss following brain damage by enhancing new synaptic connections.
Mechanisms of plasticity
Synaptic plasticity can occur at either the presynaptic or postsynaptic terminal. Modifications to the presynaptic terminal affect the release of neurotransmitters. As the action potential invades the presynaptic terminal, it activates voltage-gated calcium channels that conduct calcium ions into the presynaptic terminal. This rise in intracellular calcium triggers the exocytosis of vesicles (fusion with the plasma membrane) and thus the release of neurotransmitters. Each presynaptic terminal contains between 200 and 500 vesicles, though only a small proportion of these are ready to be released at any time. Vesicles in the presynaptic terminal move through a specific release cycle, including vesicle storage, priming for release, release, vesicle reformation, and reloading with neurotransmitter.Factors that alter the presynapse resulting in either modification of the calcium channel conductance or modification of the vesicle cycle will yield changes in synaptic strength. One such factor is the cyclic nucleotide cAMP. An increase in cAMP presynaptically can enhance transmitter release by activating protein kinase A (PKA). PKA activation induces a decrease in a specific potassium channel conductance called a delayed rectifier current. Decreased delayed rectifier conductance will increase the calcium entry into the presynaptic terminal by increasing the duration of the action potential. In addition, a rise in cAMP can activate vesicular release from presynaptic terminals that were previously dormant. Such terminals are present, but do not release neurotransmitters in response to an action potential prior to a rise in cAMP. A morphologically distinct synapse that is physiologically dormant has been termed a silent synapse and can be the result of deficient presynaptic release, or a deficiency of transmitter receptors expressed postsynaptically.
The postsynaptic terminal can also be modified to produce changes in synaptic efficacy. Signaling molecules in the postsynaptic compartment such as protein kinase A (PKA) and the alpha subunit of calcium/calmodulin-dependent kinase II (α-CaMKII) are thought to play major roles in synaptic plasticity. For example, when a mouse is genetically altered to express a version of α-CaMKII incapable of activation, LTP and learning are disrupted. While α-CaMKII can directly phosphorylate neurotransmitter receptors leading to an increase in conductance, it is likely to play additional roles in synaptic plasticity as well. Neurotransmitter receptors can cycle in and out of the postsynaptic membrane (in a process not unlike the presynaptic vesicles), and α-CaMKII phosphorylation of an as yet unidentified substrate could lead to the rapid insertion of more receptors. This would result in LTP of an active synapse and the unsilencing of a synapse that was not previously expressing these receptors in its membrane. As stated above, there is substantial evidence implicating long-lasting changes in synaptic strength with the formation of memory. It should be noted that synapses do not act in isolation. The neural circuits to which they belong are a result of the many thousands of synapses contained therein. Although the cellular coding of information may be encoded at synapses, memory itself is likely dependent upon the circuit(s) in which they are contained.
Plasticity, memory, and aging
As humans age, an impairment of memory occurs that is not associated with neurological damage or disease. The age of onset for this decline varies, but it is clear that this is a selective deficit and not a generalized decrease in cognitive skills. Moreover, the deficit is also apparent in animal models of aging and is manifest as a greater number of trials required to memorize a task and a decrease in memory retention that begins approximately twenty-four hours post-training. Interestingly, LTP also changes with age, typically requiring a more robust stimulus to induce and yielding a synaptic potentiation that decays more rapidly. Since aging animals and humans both maintain the ability to store memory, the fundamental mechanisms that underlie information storage may remain essentially intact. The deficit may not be a lack of ability, but rather a decline in the efficiency of storage—or an inability to maintain the neural plasticity induced during learning. Since the formation of memory is dependent on new protein synthesis, one way to address the decreased stability of memory is to identify proteins made during learning. Consistent with this, synaptic plasticity has at least two temporally distinct components: transient changes that do not require new protein synthesis, and enduring modifications (e.g., LTP and LTD) that require the production of new proteins. Identification of newly formed proteins, their site of action, and the molecular basis for their role in neural plasticity may provide insights into the maintenance of memory, and thus indicate clinical targets for the amelioration of age-related memory decline.David G. Wells
See also Brain; Learning; Memory; Neurochemistry; Neurodegenative Diseases.
BIBLIOGRAPHY
Bliss, T. V. P., and Lomo, T. "Long-Lasting Potentiation of Synaptic Transmission in the Dentate Area of the Anaesthetized Rabbit Following Stimulation of the Perforant Path." Journal of Physiology (London) 232 (1973): 331–356.Cowen, W. M., and Kandel, E. R. "A Brief History of Synapses and Synaptic Transmission." In Synapses. Edited by W. M. Cowen, T. C. Sudhof and C. F. Stevens, Baltimore, Md.: The Johns Hopkins University Press, 2001. Pages 1–88.
Davis, H. P., and Squire, L. R. "Protein Synthesis and Memory: A Review." Psychology Bulletin 96 (1984): 518–559.
Dudek, S. M., and Bear, M. F. "Homosynaptic Long-Term Depression in Area CA1 of Hipocampus and Effects of N-methyl-D-aspartate Receptor Blockade." Proceedings of the National Academy of Science 89 (1992): 4363–4367.
Foster, T. C. "Involvement of Hippocampal Synaptic Plasticity in Age-Related Memory Decline." Brain Research Review 30 (1999): 236–249.
Giese, K. P.; Fedorov, N. B.; Filipkowski, R. K.; and Silva, A. J. "Autophosphorylation at Thr286 of the Alpha Calcium-Calmodulin Kinase II in LTP and Learning." Science 279 (1998): 870–873.
Hayashi, Y.; Shi, S.-H.; Esteban, J. A.; Piccini, A.; Poncer, J. C.; and Malinow, R. "Driving AMPA Receptors into Synapses by LTP and CaMKII: Requirements for GluR1 and PDZ Domain Interactions." Science 287 (2000): 2262–2267.
Ma, L.; Zablow, L.; Kandel, E. R.; and Siegelbaum, S. A. "Cyclic AMP Induces Functional Presynaptic Boutons in Hippocampal CA3-CA1 Neuronal Cultures." National Neuroscience 2 (1999): 24–30.
Tong, G.; Malenka, R. C.; and Nicoll, R. A. "Long-Term Potentiation in Cultures of Single Hippocampal Granule Cells: A Presynaptic Form of Plasticity." Neuron 16 (1996): 1147–1157.
Neural plasticity: consequences of stress and actions of antidepressant treatment
This article has been cited by other articles in PMC.
Abstract
Neural
plasticity is emerging as a fundamental and critical mechanism of
neuronal function, which allows the brain to receive information and
make the appropriate adaptive responses to subsequent related stimuli.
Elucidation of the molecular and cellular mechanisms underlying neural
plasticity is a major goal of neuroscience research, and significant
advances have been made in recent years. These mechanisms include
regulation of signal transduction and gene expression, and also
structural alterations of neuronal spines and processes, and even the
birth of new neurons in the adult brain. Altered plasticity could
thereby contribute to psychiatric and neurological disorders. This
article revievi/s the literature demonstrating altered plasticity in
response to stress, and evidence that chronic antidepressant treatment
can reverse or block the effects, and even induce neural piasiicity-iike
responses. Continued elucidation of the mechanisms underlying neural
plasticity will lead to novel drug targets that could prove to be
effective and rapidly acting therapeutic interventions.
Keywords: signal transduction, gene expression, neurotrophic factor, neurogenesis, neuronal atrophy
Neural
plasticity is a fundamental process that allows the brain to receive
information and form appropriate adaptive responses to the same or
similar stimuli. The molecular and cellular adaptations underlying
learning and memory are the best-characterized and moststudied examples
of neural plasticity. However, many different stimuli can activate
neural plasticity processes in different brain structures, including
environmental, social, behavioral, and pharmacological stimuli. In fact,
it could be argued that neural plasticity is one of the most essential
and important processes that the brain performs as it relates to many
types of central nervous system functions.
Thus,
disrupted or abnormal plasticity could lead to maladaptive neuronal
responses and abnormal behavior. This could occur in response to genetic
abnormalities of the cellular machinery required for plasticity, and
abnormal or inappropriate stimuli. For example, exposure to
inappropriate or prolonged stress has been reported to alter molecular
and cellular markers of neural plasticity, and could contribute to
stress-related mood disorders. This review will discuss the literature
demonstrating altered neural plasticity in response to stress, and
clinical evidence indicating that altered plasticity occurs in depressed
patients. The second part of the review will present evidence that
antidepressant treatment blocks the effects of stress or produces
plasticity -like responses.
General mechanisms of neural plasticity
Neural
plasticity encompasses many different types of molecular and cellular
responses that occur when cells in the brain are induced to respond to
inputs from other cells or circulating factors. The systems that have
been most extensively studied are cellular and behavioral models of
learning and memory, including long-term potentiation (LTP), in slices
of brain and rodent models of behavior. The mechanisms identified for
learning and memory most likely also subserve plasticity occurring in
other regions and for other adaptive functions of the brain. This
section will briefly discuss some general mechanisms and concepts of
plasticity.
Mechanisms of acute neural plasticity: synaptic transmission and protein kinases
The
effects underlying the rapid responses to neuronal activation are
mediated by activation of the excitatory neurotransmitter glutamate and
regulation of intracellular signaling cascades (for a review of acute
mechanisms underlying LTP, see reference 1). Glutamate causes neuronal
depolarization via activation of postsynaptic ionotropic receptors that
increase intracellular Na+. This leads to the subsequent activation of
/V-mcthyl-D-aspartatc (NMDA) receptors and the resulting influx of Ca2+. Ca2+ is a major intracellular signaling molecule that activates a signaling cascade, including activation of Ca2+/ calmodulin-dependent protein kinase. Within minutes to hours, activation of glutamate and Ca2+-dependent
pathways can result in structural alterations at the level of dendritic
spines. Spines mark the location of glutamate synapses and have been
the subject of intensive investigation for understanding synaptic
plasticity.2
Changes in the shape and even number of spines can occur very rapidly
(minutes to hours) after glutamate stimulation. These alterations are
made permanent or long-term when they arc stabilized or consolidated, a
process that requires gene expression and protein synthesis.
Mechanisms of long-term plasticity: gene expression and protein synthesis
The
Ca2+/cyclic adenosine monophosphate (cAMP) response element (CaRE)
binding protein (CREB) is one of the major transcription factors that
mediate the actions of Ca2+, as well as cAMP signaling. CREB
has been reported to play a role in both cellular and behavioral models
of learning and memory.3 There are a number of gene targets that are influenced by Ca2+,
cAMP, and CREB, and the pattern of gene regulation is dependent on the
cell type, the length of stimulation, as well as the magnitude of
stimulation. Gene targets that have been implicated in learning and
memory, and are relevant to the effects of stress and antidepressant
treatment, are the neurotrophic factors. Of particular interest is
brain-derived neurotrophic factor (BDNF), one of the most abundant
neurotrophic factors in the brain.
Altered neural plasticity in response to stress
Recent
reports have demonstrated altered molecular and cellular responses to
stress and have contributed to the hypothesis that altered neural
plasticity contributes to stress-related psychiatric illnesses. Some
examples of stress responses are discussed in this section.
Stress alters learning and memory
Stress
is known to significantly influence learning and memory, and the
effects are dependent on the type, duration, and intensity of the
stressor. Emotional arousal can enhance learning and memory via synaptic
plasticity of amygdala-dependent pathways, and this is thought to be
the basis for intense, long-term memories of traumatic events and
posttraumatic stress disorder.4,5 However, stress can also impair subsequent learning and memory and can even lead to amnesia.6 The influence of stress on hippocampal-dependent learning is complex and dependent on the type of learning task.
In
studies of LTP, a consistent suppression of neural plasticity is
observed after exposure to stress or adrenal glucocorticoids.6,7
In one of these studies, the suppression of LTP was observed after
exposure to an uncontrollable stressor and correlated with behavioral
performance in a learning and memory task. Giving the animals control
over the stress (ie, the stress could be terminated) did not lead to
reduced LTP or decreased learning and memory.8 A role for BDNF in the actions of stress on LTP has also been suggested.9 For additional references and discussion of the effects of stress on learning and memory, see the reviews in references 4 to 7.
Stress causes atrophy of hippocainpal neurons
One
of the best-characterized examples of altered structural plasticity in
response to stress is the atrophy of hippocampal neurons, which was
first described by McEwen and colleagues (Figure 1.).10
They found that repeated restraint stress results in atrophy of the
dendrites of CA3 pyramidal neurons in the hippocampus, measured as a
decrease in the number and length of apical dendrites.11
The reduction in dendritic arborization was found to be dependent on
long-term, repeated exposure to restraint stress (3 weeks) and to be
reversible when the animals are removed from stress. The atrophy of CA3
pyramidal cells appears to result from the elevation of adrenal
glucocorticoids that occurs during stress because chronic administration
of corticosteronc, the active form in rodent, results in a similar
decrease in number and length of dendrites.12
The actions of stress and glucocorticoids are blocked by administration
of an NMDA receptor antagonist, indicating that this glutamate receptor
is required for atrophy of CA3 neurons.10
Atrophy of CA3 pyramidal neurons occurs after 2 to 3 weeks of exposure
to restraint stress or more long-term social stress, and has been
observed in rodents and tree shrews.11-13
In contrast to the atrophy of hippocampus, recent studies demonstrate
that chronic stress causes hypertrophy of neurons in the amygdala.14
This study found chronic immobilization stress increased the dendritic
arborization of pyramidal neurons in the basolateral amygdala, but
decreased dendrite length and branching in the CA3 pyramidal neurons of
the hippocampus. Hypertrophy of the amygdala could underlie increased
learning and memory as a result of stressinduced emotional arousal, and
may be relevant to the pathophysiology of stress-related disorders,
including anxiety, posttraumatic stress, and depression. Increased
arborization of neurons in the amygdala could thereby enhance emotional
states or disrupt normal processing of emotional responses.

Model
of hippocampal plasticity showing structural alterations in response to
stress: atropy of CA3 pyramidal neurons and decreased neurogenesis of
dentate gyrus granule cells. Stress results in powerful effects on the
hippocampus, partly because of the ...
Stress decreases neurogenesis in the adult hippocampus
In
addition to regulation of the morphology of neurons in the hippocampus,
stress influences the number of newborn neurons or neurogenesis in the
adult hippocampus15,16 (Figures 1 and 2.)
The hippocampus is one of two brain regions where neurogenesis
continues to occur in adult organism (the other region is in the
subventricular zone). In the hippocampus, neural progenitor cells are
found in the subgranular zone, between the granule cell layer and the
hilus. These cells give rise to newborn cells that migrate into the
granule cell layer and mature into neurons with the morphological and
physiological characteristics of adult granule cells.17
Interestingly, the process of neurogenesis is highly regulated by a
variety of stimuli and can be considered a form of neural plasticity.
For example, enriched environment, exercise, and learning increase
neurogenesis, while aging and exposure to drugs of abuse decrease
neurogenesis.15,16,18

Model
demonstrating the regulation of adult neurogenesis in the hippocampus.
Neural progenitor cells are restricted to the subgranular zone (SGZ)
that is located between the granule cell layer (GCL) and hilus. These
progenitor cells give rise to newborn ...
In addition to these factors, stress also results in a dramatic downrcgulation of neurogenesis in the hippocampus.10,18
Exposure to just a single stressor is sufficient to significantly
decrease neurogenesis in the adult hippocampus. Adult neurogenesis is
decreased by different types of stress, including subordination stress,19 predator odor,20 maternal separation,21 and footshock.22
In addition, exposure to inescapable stress in the learned helplessness
model of depression decreases adult neurogenesis and this effect
correlates with behavioral despair in this model.22 Moreover, the reduction in neurogenesis and the behavioral despair is reversed by antidepressant treatment.
Regulation of CREB and decreased expression of BDNF in response to stress
Stress
results in a wide range of effects that influence many different
neurotransmitter and neuropeptide systems, signal transduction pathways,
and altered gene expression. The hallmark of the stress response is
activation of the hypothalamic-pituitary-adrcnal (HPA) axis, which
includes increased circulating levels of adrenal glucocorticoids. The
hippocampus contains veryhigh levels of glucocorticoid receptors and is
therefore significantly impacted by stress. As mentioned above, studies
by McEwen and colleagues have demonstrated that glucocorticoids
contribute to the atrophy and decreased neurogenesis of hippocampal
neurons resulting from exposure to stress.10
In addition, stress is reported to influence CREB and
BDNF in the hippocampus and other brain regions. The transcriptional
activity of CREB is regulated by phosphorylation and levels of
phospho-CREB are used as an indirect measure of CREB activation and
function (Figure 3.) The, regulation of phospho-CREB is complex and is dependent on the brain region and whether the stress is acute or chronic.23-26
Acute stress increases levels of phospho-CREB in many limbic regions
associated with mood disorders and this may represent a normal or
appropriate adaptive responsiveness.24
In contrast, chronic stress leads to decreased levels of phosphoCREB in
many limbic brain regions, which could lead to decreased plasticity and
function.26

Model
demonstrating the upregulation of the cyclic adenosine monophosphate
(cAMP)-cAMP response element binding protein (CREB) cascade and
expression of brain-derived neurotrophic factor (BDNF) by antidepressant
treatment. Chronic, but not acute, antidepressant ...
Stress
has profound effects on the expression of BDNF in the hippocampus.
Levels of BDNF expression in hippocampus are dramatically downregulated
by both acute and chronic stress, and this effect could contribute to
the atrophy and decreased neurogenesis caused by stress (Figure l).27-29
The role of other factors that could underlie the actions of stress on
adult neurogenesis is a subject of interest and could lead to novel
targets for drug development.
Atrophy of limbic brain structures in depressed patients
Evidence
from basic research studies provide strong support for the hypothesis
that stress-related illnesses such as depression could include
alterations in brain structure and neural plasticity. Indeed, direct
evidence to support this hypothesis has been provided by brain imaging
and postmortem studies of depressed patients.
Evidence from brain imaging studies
Magnetic
resonance imaging studies have demonstrated that the size of certain
brain structures is decreased in mood disorder patients. In particular,
these studies demonstrate that the volume of the hippocampus is
decreased in patients with depression.30,31 Reduced hip pocampal volume is also observed in patients with posttraumatic stress disorder (PTSD).32 The reduction in hippocampal volume is directly related to the length of illness.33,34
In addition to hippocampus, atrophy of prefrontal cortex and amygdala -
brain regions that control cognition, mood, and anxiety - has also been
reported in patients with depression or bipolar disorder.35
Evidence from postmortem studies
Atrophy
of hippocampus or other brain regions could result from loss of cells
(neurons or glia) or decreased size of the cell body or neuronal
processes. The most extensive studies have been conducted on prefrontal
and cingulatc cortex and demonstrate that the neuronal body size and
number of glia is decreased in depressed patients.36-38
There is much less known about the hippocampus and additional studies
will be required to determine what accounts for the atrophy of
hippocampus observed in depressed patients.
Postmortem
analysis of CREB and BDNF has also provided evidence consistent with a
loss of neural plasticity in depression. Levels of CREB arc decreased in
the cerebral cortex of depressed patients or suicide victims.39,40 Levels of BDNF are also decreased in prefrontal cortex and hippocampus of depressed patients.41
Reduced levels of CREB and BDNF“, two molecular markers of neural
plasticity, indicate that the ability of limbic brain structures to
mount adaptive responses is compromised in depressed patients.
Antidepressant treatment increases neural plasticity
In
contrast to the effects of stress, antidepressant treatment results in
molecular and cellular responses that demonstrate an increase in neural
plasticity. Moreover, these studies have paved the way for additional
studies that demonstrate that antidepressant treatment results in
structural remodeling. In many cases, the effects of antidepressant
treatment oppose or reverse the effects of stress. Taken together, these
findings provide additional support for the hypothesis that neural
plasticity plays a significant role in the treatment, as well as the
pathophysiology of mood disorders. The evidence for regulation of neural
plasticity at the level of neurogenesis, signal transduction, and gene
expression is discussed in the second half of this review.
Antidepressant treatment increases adult neurogenesis
Neurogenesis is increased by chronic antidepressant administration
One
of the most surprising discoveries of recent times in the field of
depression is that antidepressant treatment regulates neurogenesis in
the adult hippocampus (Figures 1 and 2). In contrast to the
actions of stress, chronic antidepressant treatment increases the number
of newborn neurons in the adult hippocampus of rodents or tree shrews.42,43
The upregulation of neurogenesis is dependent on chronic antidepressant
treatment, consistent with the time course for the therapeutic action
of antidepressants.43
In addition, different classes of antidepressants, including serotonin
(5-hydroxytryptamine [5-HT]) and noradrenaline reuptake inhibitors, and
electroconvulsive seizures are reported to increase adult neurogenesis.43-45
Antidepressant treatment influences two important aspects of
neurogenesis, the rate of cell proliferation (ie, the number of newborn
neurons) and the survival of newborn neurons.46
An increase in the number of newborn neurons could contribute to the
reversal of hippocampal atrophy observed in depressed patients.
Antidepressant treatment blocks the downregulation of neurogenesis caused by stress
The
influence of antidepressant treatment in the context of stress has also
been examined. These studies demonstrate that chronic antidepressant
treatment can block or reverse the downregulation of neurogenesis that
results from exposure to stress. Several different types of stress have
been tested, including blockade of intruder stress,42 maternal separation,47 and learned helplessness.22 In addition, different types of antidepressants have been tested, including an atypical antidepressant, tianeptine,42 a selective serotonin reuptake inhibitor (SSRI),22,47 and a neurokinin-1 receptor antagonist.48.
The
influence of antidepressant treatment on the atrophy of CA3 pyramidal
neurons resulting from chronic exposure to stress has been examined.
These studies demonstrate that chronic administration of tianeptine
blocks the atrophy of CA3 apical dendrites that is caused by stress.12
Chronic administration of an SSRI antidepressant did not block the
atrophy of CA3 neurons in this study Analysis of dendrite branch number
and length is tedious and labor intensive, but additional studies of
other antidepressants are necessary to determine the relevance of this
effect in the actions of antidepressant treatment.
A functional role for neurogenesis in the action of antidepressant treatment
A
major issue in the field of adult neurogenesis is how to test the
function of newborn neurons. A recent study has addressed this question
by using a combination of irradiation and mutant mouse approaches.49
This study demonstrates that focused irradiation of hippocampus in the
mouse completely blocks neurogenesis and there was a corresponding
blockade of the behavioral actions of antidepressant treatment in two
behavioral models, novelty suppressed feeding and chronic mild stress.
In addition, Santarelli et al49 studied the effects of antidepressants in mice with a null mutation of the 5-HT1A
receptor, a subtype that has been implicated in the actions of
antidepressant treatment. They found that upregulation of neurogenesis
by chronic administration of an SSRI was completely blocked in 5-HT1A
null mutant mice, and that the behavioral effects of SSRI treatment
were similarly blocked. These results are the first evidence that
increased neurogenesis is necessary for an antidepressant response in
behavioral models. rFh ere arc a few limitations to this study. First,
although novelty-suppressed feeding is responsive to chronic
antidepressant treatment - and this is why it was chosen - this paradigm
is a better model of anxiety than depression. Second, although the
effects of antidepressant treatment were blocked, irradiation and 5-HT1A
null mutation alone, in the absence of antidepressant administration,
did not produce a depressive phenotype. This is consistent with another
report demonstrating that decreased neurogenesis is not correlated with
behavior in the learned helplessness model of depression.50
Together these studies indicate that neurogenesis is not required for
baseline response. However, it is possible that intact neurons are
sufficient to sustain baseline response and that more long-term
inhibition of neurogenesis would be required to influence activity.
The cAMP-CREB cascade and depression
Neural
plasticity upon antidepressant treatment is likely to involve
adaptations of multiple intracellular signaling cascades and even
interactions of these pathways. One of the pathways that is regulated by
antidepressant treatment and has been demonstrated to contribute to the
actions of chronic antidepressant responses is the cAMP-CREB cascade,
the subject of this section. However, it is likely that other signaling
pathways are also regulated by - and play a role in - the actions of
antidepressants. For reviews covering other signal transduction
pathways, see reference 51 and 52.
Antidepressant treatment upregulates the cAMP CREB cascade
Several studies have investigated the influence of antidepressant treatment on the cAM'P-CREB pathway (Figure 3).53,54
This work demonstrates that chronic antidepressant treatment
upregulates the cAMP second-messenger cascade at several different
levels. This includes increased coupling of the stimulatory G protein to
adenylyl cyclase, increased levels of cAMP-dependent protein kinase
(PKA), and increased levels of CREB as well as phospho-CREB.55-57
Upregulation of these components of the cAMP-CREB signaling pathway is
dependent, on chronic antidepressant treatment, consistent with the time
course for the therapeutic action of antidepressants. In addition,
upregulation of the cAMP-CREB cascade is observed in response to chronic
administration of different classes of antidepressants, indicating that
this is a common target of antidepressant treatment.
In addition to phosphorylation by PKA, CREB is also phosphorylated by Ca2+-dependent kinases, such as Ca2+/calmodulin-dependent protein kinase, and by mitogen-activated protein kinase pathways (Figure 3).
In this way, CREB can serve as a target for multiple signal
transduction pathways and neurotransmitter receptors that activate these
cascades.
Activation of the cAMP-CREB cascade produces an antidepressant response
Direct,
evidence for cAMP-CREB signaling in the action of antidepressant
treatment has been tested by pharmacological, viral vector, and mutant
mouse approaches. First, drugs that block the breakdown of cAMP produce
an antidepressant response in behavioral models of depression.54
The primary target for inhibition of cAMP breakdown is cAMP-specific
phosphodiesterase type IV (PDE4), and rolipram was one of the first
selective PDE4 inhibitors. In addition, we have found that chronic
rolipram administration increases neurogenesis in adult hippocampus.46,58
Second, viral expression of CREB in
the hippocampus of rat produces an antidepressant response in the forced
swim and learned helplessness models of depression.59
However, further studies demonstrated that the effects of CREB are
dependent on the brain region where it is expressed. For example,
expression of CREB in the nucleus accumbens produces a prodepressant
effect, while expression of a dominant, negative mutant of CREB results
in an antidepressant response in the forced swim test.60 Transgenic expression of dominant negative CREB in the nucleus accumbens is consistent with this effect.61
The different behavioral effects of CREB can be explained by different
target genes in the hippocampus (ic, BDNF) versus the nucleus accumbens
(ie,prodynorphin).
Regulation of neurotrophic factors and depression
The
regulation of CREB by antidepressant treatment indicates that
regulation of gene expression also plays a role in the actions of
antidepressants. There have been many gene targets identified for
antidepressants,51,52
but BDNF is one that has gained attention and is relevant to neural
plasticity responses to antidepressant medications. Studies to identify
additional gene targets and gene profiles using gene microarray analysis
are currently being conducted.
Antidepressant treatment upregulates BDNF
Neurotrophic
factors were originally identified and studied for their role in
development, and neuronal survival. However, it is now clear that these
factors are expressed in the adult brain, arc dynamically regulated by
neuronal activity, and are critical for the survival and function of
adult neurons. On the basis of these considerations, it is clear why
decreased expression of BDNF could have serious consequences for the
function of limbic brain structures that control mood and cognition. In
contrast, antidepressant treatment results in significant upregulation
of BDNF in the hippocampus and cerebral cortex of rodents.28,53,54
Increased expression of BDNF is dependent on chronic treatment, and is
observed with different classes of antidepressants, but not other
psychotropic drugs. The induction of BDNF would be expected to protect
neurons from damage resulting from stress, elevated glucocorticoids, or
other types of neuronal insult.
BDNF has antidepressant effects in behavioral models of depression
The
possibility that BDNF contributes to the actions of antidepressant
treatment is supported by behavioral studies of recombinant BDNF and
transgenic mouse models. Microinfusions of BDNF into the hippocampus
produce an antidepressant-like response in the learned helplessness and
forced swim models of depression.62
The antidepressant, effect of BDNF is observed after a single infusion,
compared with repeated administration of a. chemical antidepressant,
and is relatively long-lasting (up to 10 days after infusion).
Transgenic overexpression of a dominant negative mutant of the BDNF
receptor, trkB, in the hippocampus and other forebrain structures is
also reported to block the effect, of antidepressant treatment,
demonstrating that BDNF signaling is necessary for an antidepressant
response.63
Microinfusions of BDNF into the
dorsal raphe, a midbrain region where 5-HT cell bodies are localized,
also produces an antidepressant response in the learned helplessness
model.64
Together, these studies indicate that BDNF could contribute to
antidepressant responses in both forebrain and brain stem structures by
affecting different populations of neurons. Alternatively, it is
possible that, microinfusions of BDNF into the hippocampus influence
5-HT neuronal function by acting at presynaptic sites, and could
therefore enhance 5-HT signaling as observed after brain stem infusions
of BDNF.64
A neurotrophic hypothesis of depression
Basic research and clinical studies of BDNF have resulted in a. neurotrophic hypothesis of depression and antidepressant action.53,54
This hypothesis is based in part. on studies demonstrating that stress
decreases BDNF, reduces neurogenesis, and causes atrophy or CA3
pyramidal neurons. Brain imaging and postmortem studies provide
additional support, demonstrating atrophy and cell loss of limbic
structures, including the hippocampus, prefrontal cortex, and amygdala.
In contrast, antidepressant treatment, opposes these effects of stress
and depression, increasing levels of BDNF, increasing neurogenesis, and
reversing or blocking the atrophy and cell loss caused by stress and
depression. Additional brain imaging and postmortem studies, as well as
basic research approaches will be required to further test this
hypothesis. In any case, the studies to date provide compelling evidence
that, neural plasticity is a. critical factor in the pathophysiology
and treatment of depression.
Antidepressants influence other neurotrophic factor systems
Because
of the preclinical and clinical evidence implicating neurotrophic
factors in the pathophysiology and treatment of depression, studies have
been conducted to examine other neurotrophic factor systems. One of the
most robust effects identified to date is that antidepressant treatment
increases the expression of fibroblast. growth factor-2 (FGF-2).65
FGF-2 is known to have a potent influence on neurogenesis during
development and in the adult brain, and could contribute to antide
pressant regulation of neurogenesis. Studies are under way to examine
the role of FGF-2 in antidepressant regulation of neurogenesis and
regulation of behavior in models of depression. Several other growth
factors have been identified by microarray analysis and gene expression
profiling, including vascular endothelial growth factor, neuritin, and
VGF.66 Studies are currently under way to determine the functional significance of these growth factors in models of depression.
Clinical evidence of relevance of neural plasticity to antidepressant treatment
Basic
research studies clearly demonstrate that antidepressant treatment
regulates signal transduction, gene expression, and the cellular
responses that, represent neural plasticity. This issue is more
difficult, to address in clinical studies, but evidence is slowly
accumulating. Brain imaging studies have been conducted to examine the
influence of antidepressants on the volume of limbic brain regions. One
study demonstrates that hippocampal atrophy is inversely proportional to
the length of time a patient receives antidepressant medication.67
A longitudinal study of PTSD patients before and after antidepressant
treatment has found that there is a. partial reversal of hippocampal
atrophy in patients receiving medication.68 The latter study demonstrated a corresponding increase in verbal declarative memory in response to antidepressant treatment.
Evidence
at the molecular level is also provided by postmortem studies. Levels
of CREB immunoreactivity are increased in patients receiving
antidepressant treatment at the time of death relative to unmedicated
patients.39 In addition, levels of BDNF are increased in patients taking an antidepressant at the time of death.59
Although these effects must be replicated and extended (for example, to
the regulation of neurogenesis) in additional banks of postmortem
tissue, the results are consistent with the hypothesis that neural
plasticity is upregulatcd in patients receiving antidepressant
medication.
Novel targets for the treatment of depression
The hypothesis that antidepressant
treatment increases neural plasticity provides a number of novel targets
for drug development. However, as with any fundamentally important
mechanism, care must be taken that the drugs developed for such targets
do not interfere with the normal function of the brain. Nevertheless,
regulation of neural plasticity is an exciting area of research for
design of new drugs for a variety of indications, including learning,
memory, cognition, mood, and neurodegenerative disorders. This section
discusses a few of these targets in the context of the pathways
regulated by antidepressants and stress.
Targets for antidepressant regulation of neurogenesis
Identification
of the signal transduction and gene expression pathways that are
responsible for the actions of antidepressant regulation of neurogenesis
is a subject, of intense investigation. Activation of the cAMP-CREB
signaling cascade using either pharmacological or transgenic approaches
is reported to increase both proliferation and survival of newborn
neurons in the hippocampus,46,58
supporting the possibility that antidepressants increase neurogenesis
via regulation of this intracellular pathway. Gene targets of CREB, as
well as other neurotrophic/growfh factors that, have been shown to
regulate adult neurogenesis, include BDNF, FGF-2, and insulin-like
growth factor-1 , to name but. a few.18
Because antidepressant treatment increases the expression of both BDNF
and FGF-2, these two factors are currently being investigated. This is
just a partial listing of the signal transduction cascades and factors
that could contribute to antidepressant regulation of adult
neurogenesis.
Targets for regulation of the cAMP-CREB cascade
There
are several different sites within the cAMP pathway that could be
targeted for drug development. One that has already proven to be
effective for antidepressant treatment is blockade of PDE4 and the
breakdown of cAMP. Rolipram is a PDF'4-selective inhibitor that has been
demonstrated to have antidepressant efficacy in early clinical trials
and behavioral models of depression.69,70 However, the clinical use of rolipram has been limited by its side effects, primarily nausea.
The
identification of four different. PDE4 isozymes that are equally
inhibited by rolipram raises the possibility that one of the isozymes
underlies the antidepressant actions of rolipram, while another mediates
its side effects. Studies are currently under way to characterize the
regional distribution and function of the three PDE4 isozymes expressed
in brain (PDE4A, PDE4B, and PDE4D) and the role of these isozymes in the
actions of antidepressant treatment.71
Studies of mutant mice demonstrate that null mutation of PDE4D produces
an antidcpressant-like phenotype indicating a role for this isozyme,72 and similar studies are currently under way for PDE4A and PDE4B.
BDNF as a target for drug development
The
use of BDNF and other neurotrophic factors for the treatment of
neurological disorders has been a subject of interest, for several
years, although problems with delivery, efficacy, and side effects have
hampered these efforts. To more directly replicate the in vivo
situation, it may be possible to stimulate the expression of endogenous
BDNF expression by stimulating signaling pathways known to regulate this
neurotrophic factor. First, activation of the cAMP-CREB cascade by
inhibition of PDE4 increases the expression of BDNF.56
Small molecular agonists for
neurotransmitter receptors have also exhibited some promise. Activation
of ionotropic glutamate receptors increases BDNF expression and could be
targeted for the treatment of depression.73 One drug that modulates glutamate transmission and increases BDNF expression is memantine.74 Riluzole, a. sodium channel blocker, also increases BDNF expression, as well as neurogenesis in adult hippocampus.75 Specific 5-HT and norepinephrine receptor subtypes that activate cAMP (eg, β-adrenergic, 5-HT7), Ca2+, or mitogen-activated protein kinase (α1-adrenergic, 5-HT1A)
pathways could also be targets for development. Characterization of the
antidepressant actions of these compounds will be needed, as well as
identification of additional neurotransmitter and signal transduction
systems that regulate BDNF
Conclusions
Studies
of the molecular and cellular mechanisms underlying neural plasticity
responses in learning and memory, as well as fear, anxiety, depression,
and drug abuse to name but a few, are some of the most exciting and
rapidly advancing areas of research in neuroscience. Progress in our
understanding of neural plasticity has profound implications for the
treatment of a number of psychiatric and neurodegenerative disorders,
and for enhancing performance in what are considered normal subjects.
One of the promising aspects of neural plasticity is that it implies
that the alterations that occur are reversible, even neuronal atrophy
and cell loss. Reversibility of structural as well as functional
plasticity has already been demonstrated in response to pharmacological
treatments or even behavioral therapy. As the fundamental mechanisms of
neural plasticity are further elucidated, new targets and paradigms for
enhancing plasticity will be revealed and will lead to more effective
and faster-acting therapeutic interventions.
Selected abbrewiations and acronyms
| BDNF | brain-derived neurotrophic factor |
| cAMP | cyclic adenosine monophosphate |
| CaRE | cAMP response element |
| CREB | cAMP response element binding protein |
| FGF-2 | fibroblast growth factor-2 |
| 5-HT | 5 -hydroxy tryptamine (serotonin) |
| LTP | long-term potentiation |
| NMDA | N-methyl-D-aspartate |
| PDE4 | phosphodiesterase type IV |
| PKA | protein kinase |
| SSRI | selective serotonin reuptake inhibitor |
Notes
This
work is supported by USPHS grants MH45481 and 2 P01 MH25642, a Veterans
Administration National Center Grant for posttraumatic stress disorder,
and by the Connecticut Mental Health Center.
REFERENCES
1. Malenka R., Nicoll RA. Long-term potentiation - a decade of progress?. Science. 1999;285:1870–1874. [PubMed]
2. Lamprecht R., LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. [PubMed]
3. Silva A., Kogan JH., Frankland PW., Kida S. CREB and memory. Ann Rev Neurosci. 1998;21:127–148. [PubMed]
4. Cahill L., McGaugh JL. Mechanisms of emotional arousal and lasting declarative memory. Trends Neurosci. 1998;21:294–299. [PubMed]
5. LeDoux J. Emotion circuits in the brain. Ann Rev Neurosci. 2000;23:155–184. [PubMed]
6. Kim J., Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. [PubMed]
7. Pavlides C., Nivon LG., McEwen BS. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 2002;12:245–257. [PubMed]
8. Shors
T., Seib TB., Levine S., Thompson RF. Inescapable versus escapable
shock modulates long-term potentiation in the rat hippocampus. Science. 1989;244:224–226. [PubMed]
9. Zhou
J., Zhang F., Zhang Y. Corticosterone inhibits generation of longterm
potentiation in rat hippocampus slice: involvement of brain-derived
neurotrophic factor. Brain Res. 2000;885:182–191. [PubMed]
10. McEwen B. Stress and hippocampal plasticity. Curr Opin Neurobiol. 1999;5:205–216. [PubMed]
11. Wooley
CS., Gould E., McEwen BS. Exposure to excess glucocorticoids alters
dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531:225–231. [PubMed]
12. Watanabe
Y., Gould E., Daniels DC., Cameron H., McEwen BS. Tianeptine attenuates
stress-induced morphological changes in the hippocampus. Eur J Pharmacol. 1992;222:157–162. [PubMed]
13. Margarinos
A., McEwen BS., Flugge G., Fuchs E. Chronic psychosocial stress causes
apical dendritic atrophy of hippocampal CA3 pyramidal neurons in
subordinate tree shrews. J Neurosci. 1996;16:3534–3540. [PubMed]
14. Vyas
A., Mitra R., Shankaranarayana Rao BS., Chattarji S. Chronic stress
induces contrasting patterns of dendritic remodeling in hippocampal and
amygdaloid neurons. J Neurosci. 2002;22:6810–6818. [PubMed]
15. Gage F. Mammalian neural stem cells. Science. 2000;287:1433–1438. [PubMed]
16. Gould E., Beylin A., Tanapat P., Reeves A., Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–265. [PubMed]
17. van
Praag H., Schlinder AF., Christie BR., Toni N., Palmer TD., Gage FH.
Functional neurogenesis in the adult mouse dentate gyrus. Nature. 2002;415:1030–1034. [PubMed]
18. Duman R., Malberg J., Nakagawa S. Regulation of adult neurogenesis by psychotropic drugs and stress. J Pharmacol Exp Ther. 2001;299:401–407. [PubMed]
19. Gould
E., McEwen BS., Tanapat P., Galea LAM., Fuchs E. Neurogenesis in the
dentate gyrus of the adult tree shrew is regulated by psychosocial
stress and NMDA receptor activation. J Neurosci. 1997;17:2492–2498. [PubMed]
20. Tanapat
P., Hastings NB., Rydel TA., Galea LAM., Gould E. Exposure to fox odor
inhibits cell proliferation in the hippocampus of adult rats via an
adrenal hormone-dependent mechanism. J Cornp Neurol. 2001;437:496–504. [PubMed]
21. Lee K., Lynch KR., Nguyen T., et al. Cloning and charactization of additional members of the G protein-coupled receptor family. Biochim Biophys Acta. 2000;1490:311–323. [PubMed]
22. Malberg
J., Duman RS. Cell proliferation in adult hippocmpus is decreased by
inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology. 2003;28:1562–1571. [PubMed]
23. Barrot
M., Olivier JD., Perrotti LI., et al. CREB activity in the nucleus
accumbens shell controls gating of behavioral responses to emotional
stimuli. Proc Natl Acad Sci USA. 2002;99:11435–11440. [PMC free article] [PubMed]
24. Bilang-Bleuel
A., Rech J., De Carli S., Holsboer F., Reul JMHM. Forced swimming
evokes a biphasic response in CREB phosphorylation in extrahypothalamic
limbic and neocortical brain structures in the rat. Eur J Neurosci. 2002;15:1048–1060. [PubMed]
25. Bruijnzeel
A., Stam R., Compaan JC., Wiegant VM. Stress-induced sensitization of
CRH-ir but not P-CREB-ir responsivity in the rat central nervous system.
Brain Res. 2001;908:187–196. [PubMed]
26. Trentani
A., Kuipers SD., Ter Horst GJ., Den Boer JA. Selective chronic
stress-induced in vivo ERK1/2 hyperphosphorylation in medial
prefrontocortical dendrites: implications for stress-related cortical
pathology?. Eur] Neurosci. 2002;15:1681–1691. [PubMed]
27. Duman R. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromol Med. 2004;5:11–26. [PubMed]
28. Nibuya
M., Morinobu S., Duman RS. Regulation of BDNF and trkB mRNA in rat
brain by chronic electroconvulsive seizure and antidepressant drug
treatments. J Neurosci. 1995;15:7539–7547. [PubMed]
29. Smith
MA., Makino S., Kvetnansky R., Post RM. Stress alters the express of
brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the
hippocampus. J Neurosci. 1995;15:1768–1777. [PubMed]
30. Bremner J., Narayan M., Anderson ER., Staib LH., Miller H., Charney DS. Smaller hippocampal volume in major depression. Am J Psychiatry. 2000;157:115–117. [PubMed]
31. Sheline Y., Wany P., Gado MH., Csernansky JG., Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. 1996;93:3908–3913. [PMC free article] [PubMed]
32. Bremner
JD., Randall P., Scott TM., et al. MRI-based measurement of hippocampal
volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry. 1995;152:973–981 . [PMC free article] [PubMed]
33. MacQueen
G., Campbell S., McEwen BS., et al. Course of illness, hippocampal
function, and hippocampal volume in major depression. Proc Natl Acad Sci USA. 2003;100:1387–1392. [PMC free article] [PubMed]
34. Sheline
Y., Sanghavi M., Mintun MA., Gado MH. Depression duration but not age
predicts hippocampal volume loss in medically healthy wormen with
recurrent major depression. J Neurosci. 1999;19:5034–5043. [PubMed]
35. Manji
H., Duman RS. Impairments of neuroplasticity and cellular resilience in
severe mood disorders: implications for the development of novel
therapeutics. Psychopharmacol Bull. 2001;35:5–49. [PubMed]
36. Cotter
D., Mackay D., Landau S., Kerwin R., Everall I. Reduced glial cell
density and neuronal size in the anterior cingulate cortex in major
depressive disorder. Arch Gen Psychiatry. 2001;58:545–553. [PubMed]
37. Ongur D., Drevets WC., Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95:13290–13295. [PMC free article] [PubMed]
38. Rajkowska
G., Miguel-Hidalgo JJ., Wei J., et al. Morphometric evidence for
neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. [PubMed]
39. Dowlatshahi
D., MacQueen GM., Wang JF., Young LT. Increased temporal cortex CREB
concentrations and antidepressant treatment in major depression. Lancet. 1998;352:1754–1755. [PubMed]
40. Dwivedi
Y., Rizavi HS., Conley RR., Tamminga CA., Pandey GN. Altered gene
expression of brain-derived neurotrophic factor and receptor tyrosine
kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60:804–815. [PubMed]
41. Dwivedi
Y., Rizavi HS., Roberts RC., Conley RC., Tamminga CA., Pandey GN.
Reduced activation and expression of ERK1/2 MAP kinase in the postmortem
brain of depressed suicide subjects. J Neurochem. 2001;77:916–928. [PubMed]
42. Czeh
B., Michaelis T., Watanabe T., et al. Stress-induced changes in
cerebral metabolites, hippocampal volume, and cell proliferation are
prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci USA. 2001;98:12796–12801. [PMC free article] [PubMed]
43. Malberg J., Eisch AJ., Nestler EJ., Duman RS. Chronic antidepressant treatment increases neurogenesis in adult hippocampus. J Neurosci. 2000;20:9104–9110. [PubMed]
44. Madsen
T., Treschow A., Bengzon J., Bolwig TG., Lindvall O., Tingstrôm A.
Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry. 2000;47:1043–1049. [PubMed]
45. Manev
H., Uz T., Smalheiser NR., Manev R. Antidepressants alter cell
proliferation in the adult brain in vivo and in neural cultures in
vitro. Eur J Pharmacol. 2001;411:67–70. [PubMed]
46. Nakagawa
S., Kim JE., Lee R., et al. Regulation of neurogenesis in adult mouse
hippocampus by cAMP and cAMP response element-binding protein. J Neurosci. 2002;22:9868–9876. [PubMed]
47. Lee
H., Kim JW., Yim SV., et al. Fluoxetine enhances cell proliferation and
prevents apoptosis in dentate gyrus of maternally separated rats. Mot Psychiatry. 2001;6:725–728. [PubMed]
48. van
der Hart M., Czeh B., de Biurrun G., et al. Substance P receptor
antagonist and clomipramine prevent stress-induced alterations in
cerebral metabolites, cytogenesis in the dentate gyrus and hippocampal
volume. Mol Psychiatry. 2002;7:933–941. [PubMed]
49. Santarelli L., Saxe M., Gross C., et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. [PubMed]
50. Vollmayr
B., Simonis C., Weber S., Gass P., Henn F. Reduced cell proliferation
in the dentate gyrus is not correlated with the development of learned
helplessness. Biol Psychiatry. 2003;54:1035–1040. [PubMed]
51. Manji H., Drevets WC., Charney DS. The cellular neurobiology of depression. Nat Med. 2001;7:541–547. [PubMed]
52. Nestler E., Barrot M., DiLeone RJ., Eisch AJ., Gold SJ. Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. [PubMed]
53. Duman R., Heninger GR., Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. [PubMed]
54. Duman R., Malberg J., Nakagawa S. C D'Sa. Neuronal plasticity and survival in mood disorders. Biol Psychiatry. 2000;48:732–739. [PubMed]
55. Nestler
E., Terwilliger RZ., Duman RS. Chronic antidepressant administration
alters the subcellular distribution of cAMP-dependent protein kinase in
rat frontal cortex. J Neurochem. 1989;53:1644–1647. [PubMed]
56. Nibuya
M., Nestler EJ., Duman RS. Chronic antidepressant administration
increases the expression of cAMP response element binding protein (CREB)
in rat hippocampus. J Neurosci. 1996;16:2365–2372. [PubMed]
57. Thome
J., Sakai N., Shin KH., et al. cAMP response element-mediated gene
transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. [PubMed]
58. Nakagawa
S., Kim JE., Lee R., Chen J., Fujioka T., Malberg J. Localization of
phosphorylated cAMP response element-binding protein in immature neurons
of adult hippocampus. J Neurosci. 2002;22:9868–9876. [PubMed]
59. Chen
A-H., Shirayama Y., Shin KH., Neve RL., Duman RS. Expression of the
cAMP response element binding protein (CREB) in hippocampus produces
antidepressant effect. Biol Psychiatry. 2001;49:753–762. [PubMed]
60. Pliakas
A., Carlson RR., Neve RL., Konradi C., Nestler EJ., Carlezon WA.
Altered responsiveness to cocaine and increased immobility in the forced
swim test associated with elevated CREB expression in the nucleus
accumbens. J Neurosci. 2001;21:7397–7403. [PMC free article] [PubMed]
61. Newton
S., Thome J., Wallace TL., et al. Inhibition of cAMP response
element-binding protein or dynorphin in the nucleus accumbens produces
an antidepressant-like effect. J Neurosci. 2002;24:10883–10890. [PubMed]
62. Shirayama
Y., Chen AC., Nakagawa S., Russell RS., Duman RS. Brain-derived
neurotrophic factor produces antidepressant effects in behavioral models
of depression. J Neurosci. 2002;22:3251–3261. [PubMed]
63. Saarelainen
T., Hendolin P., Lucas G., et al. Activation of the trkB neurotrophin
receptor is induced by antidepressant drugs and is required for
antidepressant-induced behavioral. effects. J Neurosci. 2003;23:349–357. [PubMed]
64. Siuciak JA., Lewis DR., Wiegand SJ., Lindsay R. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav. 1997;56:131–137. [PubMed]
65. Mallei
A., Shi B., Mocchetti I. Antidepressant treatments induce the
expression of basic fibroblast growth factor in cortical and hippocampal
neurons. 2002;61:1017–1024. [PubMed]
66. Newton
S., Collier E., Hunsberger J., Adams D., Salvanayagam E., Duman RS.
Gene profile of electroconvulsive seizures: induction of neurogenic and
angiogenic factors. J Neurosci. 2003;23:10841–10851. [PubMed]
67. Sheline Y., Gado MH., Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry. 2003;160:1–3. [PubMed]
68. Vermetten
E., Vythilingam M., Southwick SM., Charney DS., Bremner JD. Long-term
treatment with paroxetine increases verbal declarative memory and
hippocampal volume in posttraumatic stress disorder. Biol Psychiatry. 2003;54:693–702. [PMC free article] [PubMed]
69. Horowski
R., Sastre-Y-Hernandez M. Clinical effects of the neurotrophic
selective cAMP phosphodiesterase inhibitor rolipram in depressed
patients: global evaluation of the preliminary reports. CurrTher Res. 1985;38:23–29.
70. Wachtel
H. Potential antidepressant activity of rolipram and other selective
cyclic adenosine 3',5“-monophosphate phosphodiesterase inhibitors. Neuropharmacology. 1983;22:267–272. [PubMed]
71. Takahashi
M., Terwilliger R., Lane S., Mezes PS., Conti M., Duman RS. Chronic
antidepressant administration increases the expression of cAMP
phosphodiesterase 4A and 4B isoforms. J Neurosci. 1999;19:610–618. [PubMed]
72. Zhang
H-T., Huang Y., Jin SJC., et al. Antidepressant-like profile and
reduced sensitivity to rolipram in mice deficient in the PDE4D
phosphodiesterase enzyme. Neuropsychopharmacology . 2002;27:587–595. [PubMed]
73. Li
X., Tizzano JP., Griffey K., Clay M., Lindstron T., Skolnick P.
Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology. 2001;40:1028–1033. [PubMed]
74. Marvanova
M., Lakso M., Pirhonen J., Nawa H., Wong G., Castren E. The
neuroprotective agent memantine induces brain-derived neurotrophic
factor and trkB receptor expression in rat brain. Mol Cell Neurosci. 2001;18:247–258. [PubMed]
75. Katoh-Semba
R., Asano T., Ueda H., et al. Riluzole enhances expression of
brain-derived neurotrophic factor with consequent proliferation of
granule precursor cells in the rat hippocampus. FASEBJ. 2001;16:1328–1330. [PubMed]






























{kind=link}